- 品牌
- 同顺生物
从作用机制看,诺拉曲特通过不可逆结合TS酶的辅因子结合位点,阻断胸腺嘧啶核苷酸合成,直接切断DNA复制的关键原料供应。与传统抗代谢药如5-氟尿嘧啶不同,其设计避免了谷氨酸侧链结构,从而消除了细胞内聚谷氨酸化导致的长期骨髓抑制风险。临床前研究显示,该药物对肝疾病HepG2细胞系的IC₅₀值为0.32μM,明显低于索拉非尼的1.2μM;在裸鼠肝疾病移植模型中,连续给药21天后疾病体积缩小67%,且未观察到肝肾功能指标异常。这种选择性毒性源于疾病细胞对TS酶的高依赖性——快速增殖的疾病细胞需要持续合成DNA,而正常细胞可通过补救途径获取胸腺嘧啶,这种代谢差异构成了诺拉曲特的医治窗口。原料药是药品生产的重要成分,直接影响药品的质量和疗效。安徽阿维巴坦
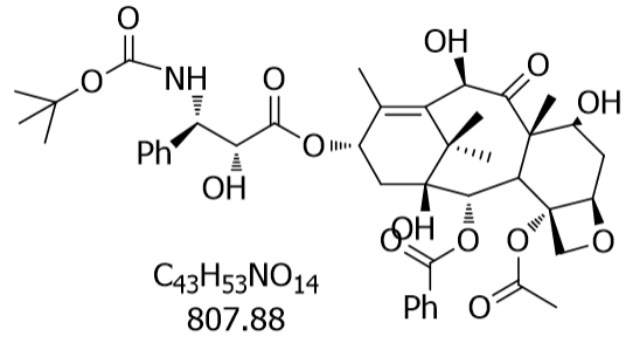
紫杉醇(Paclitaxel,CAS:33069-62-4)作为天然抗疾病药物的标志,其重要性能源于独特的分子结构与作用机制。该化合物分子式为C₄₇H₅₁NO₁₄,分子量853.91,属于二萜生物碱类化合物,其结构中包含一个由6-8-6-4环系构成的紫杉烯骨架,以及一个含氧四元环结构。这一特殊构型使其成为已知能够直接结合聚合态微管蛋白的药物。研究表明,紫杉醇通过特异性识别微管蛋白的N端区域,诱导微管蛋白二聚体聚合成稳定微管束,同时抑制微管解聚,导致细胞内微管网络异常积聚。这种作用模式不同于传统微管抑制剂(如长春碱类),后者主要作用于游离微管蛋白二聚体。临床前研究显示,紫杉醇在0.1-1μM浓度下即可明显抑制乳腺疾病细胞系CCRF-HSB-2的增殖,IC50值达0.25μM,其作用强度是长春新碱的3-5倍。此外,该药物还能通过启动内质网钙离子通道,诱导细胞凋亡信号通路,形成双重抗疾病机制。常州苏尼替尼仿制药企业采购原料药时,需确保与原研药质量一致性。
从红豆杉树皮到临床制剂,紫杉醇的工业化生产经历了从天然提取到生物合成的技术变革。传统提取法需消耗大量树皮资源,每吨树皮只能获得约0.01%的紫杉醇,导致野生红豆杉濒临灭绝。2024年,中国科学家闫建斌团队通过解析紫杉烷氧杂环丁烷合酶和T9αH酶的催化机制,在酵母中重构了巴卡亭Ⅲ(紫杉醇关键前体)的合成途径,使产量提升至10-30μg/g,达到天然红豆杉针叶的含量水平。与此同时,斯坦福大学团队利用单细胞转录组学技术,在重建了包含17个基因的完整合成通路,实现了巴卡亭Ⅲ的异源表达。这些突破不仅解决了资源瓶颈,更揭示了紫杉醇生物合成的模块化特征:早期萜烯合成模块负责碳骨架构建,中游氧化模块完成羟基化修饰,后期修饰模块进行乙酰化/去乙酰化反应。
地拉罗司不仅在医治铁过载症方面具有明显疗效,其安全性和耐受性也得到了普遍的认可。作为一种新型的三价铁螯合剂,地拉罗司能够与体内过多的铁离子高度选择性结合,形成可溶性复合物,从而帮助排出多余的铁。这种药物在Ⅱ、Ⅲ期临床试验及药代动力学研究中均表现出了良好的安全性和耐受性。它的使用可以有效控制铁过载,减轻患者的症状。除了主要的医治功能外,地拉罗司还具有一些其他的药物学特性,如抗细菌、抗细胞增殖、抗疟疾和抗氧化应激损伤等。这些特性使得地拉罗司在医治一些其他疾病方面也具有潜力。尽管地拉罗司具有诸多优点,但在使用前仍需告知医生有关自身的过敏史、现有的疾病以及正在服用的其他药物,以确保用药安全。同时,儿童、孕妇、哺乳期妇女以及肝功能受损患者在使用时需要额外谨慎。不同类型原料药需采用专属提取工艺,保障有效成分稳定留存。
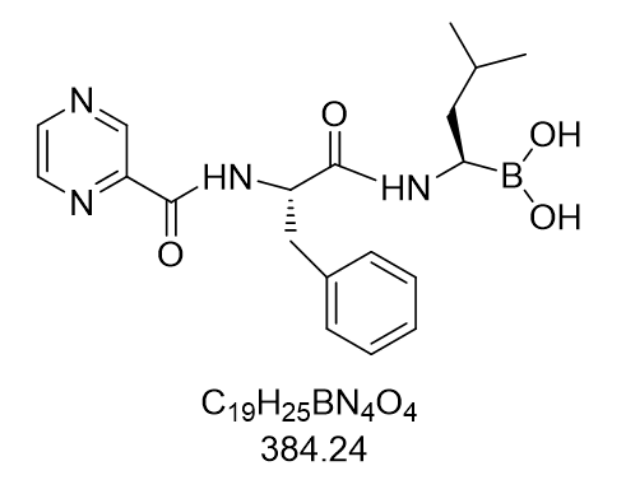
在临床前安全性评估中,德兰佐米展现出优于传统蛋白酶体抑制剂的毒性特征。急性毒性实验显示,大鼠LD50为120 mg/kg,是硼替佐米(45 mg/kg)的2.7倍。亚慢性毒性研究(28天重复给药)表明,15 mg/kg剂量组只出现轻度可逆性血小板减少(下降28%),而同等暴露量下硼替佐米会导致45%的血小板减少和30%的神经毒性发生率。机制研究发现,德兰佐米对正常细胞蛋白酶体的抑制强度只为疾病细胞的1/5,这种差异源于疾病细胞高表达的免疫蛋白酶体(i-proteasome)对其具有更高亲和力。在原代人外周血单核细胞(PBMC)中,40 nM浓度处理72小时未诱导明显凋亡,而相同条件下MM细胞存活率低于5%。更关键的是,德兰佐米不抑制T细胞蛋白酶体活性,反而通过下调PD-L1表达增强T细胞杀伤功能,这种免疫调节特性使其在联合免疫检查点抑制剂医治中具有潜在优势。目前,该化合物已进入II期临床试验(NCT03672218),针对复发/难治性MM患者的初步数据显示,客观缓解率达58%,3级以上不良反应发生率较硼替佐米方案降低40%,凸显其临床转化价值。冷冻干燥技术使原料药含水量低于0.5%,明显提升长期稳定性。沙库比曲缬沙坦钠厂家直供
原料药市场波动受供需关系与政策影响较为明显。安徽阿维巴坦
诺拉曲特(Nolatrexed,CAS号:147149-76-6)作为一款靶向胸苷酸合成酶(TS)的小分子抑制剂,其研发历程与分子机制体现了现代药物设计的精确性。该药物由美国Agouron公司通过计算机辅助药物设计技术(CADD)开发,基于TS酶的三维晶体结构,设计出能同时占据酶活性中心两个叶酸结合位点的喹唑啉衍生物结构。其分子式为C₁₄H₁₂N₄OS,分子量284.34,水溶性特性使其口服给药后生物利用度可达90%以上,且通过被动扩散穿透细胞膜,避免了传统叶酸类似物依赖转运蛋白的耐药风险。1999年Agouron将全球疾病适应症权益转让给Eximias制药后,该药物进入全球多中心临床试验阶段,其中肝疾病Ⅲ期研究因中国肝疾病高发背景成为重点布局领域,而结肠疾病、肺疾病等Ⅱ期试验则覆盖欧美12国,累计入组超2000例患者。安徽阿维巴坦
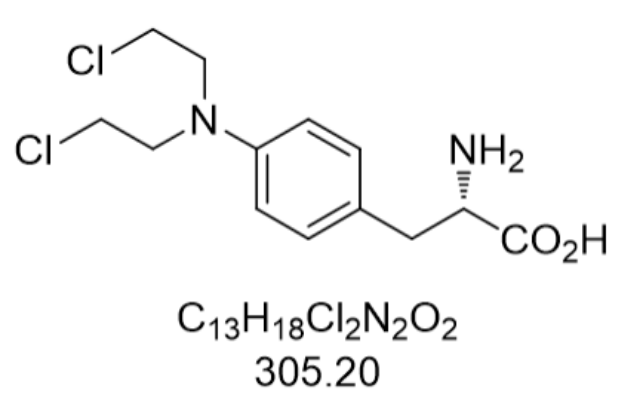
苯丁酸氮芥(Chlorambucil,CAS:305-03-3)作为经典的烷化剂类抗疾病药物,其化学本质为4-[双(2-氯乙基)氨基]苯丁酸,分子量304.212,外观呈米色结晶性粉末。该药物通过DNA烷基化作用形成交联结构,抑制疾病细胞增殖,属于细胞周期非特异性的药物。其作用机制的重要在于双功能烷化基团与DNA鸟嘌呤碱基的N7位点结合,形成链内或链间交联,阻碍DNA复制和转录。相较于其他氮芥类药物,苯丁酸氮芥的丙酸侧链在β-位氧化生成氮芥后,脱氯乙基作用缓慢,延长了药物在体内的活性时间。临床应用中,该药物对慢性淋巴细胞白血病(CLL)的缓解率可达60%-70%,尤其适用于老年或合并症较多的患...
- 沙库比曲缬沙坦钠供应公司 2026-01-26
- 诺拉曲特哪里有卖 2026-01-25
- 西安紫杉醇 2026-01-24
- 山东沙库比曲缬沙坦钠 2026-01-24
- 四川多西他赛 2026-01-23
- 北京苯丁酸氮芥 2026-01-23
- 苏尼替尼销售 2026-01-22
- 上海多西他赛 2026-01-22
- 安徽阿维巴坦 2026-01-21
- 湖北地拉罗司 2026-01-21

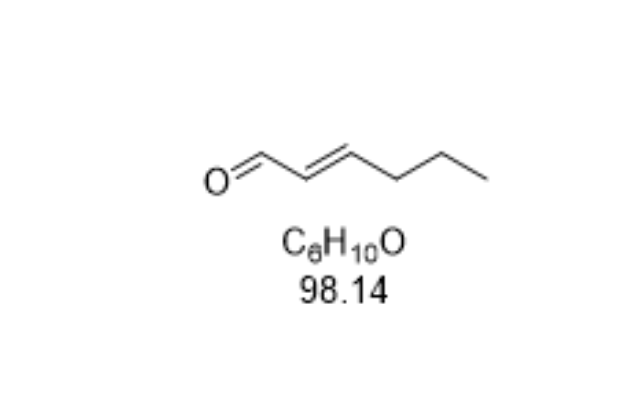
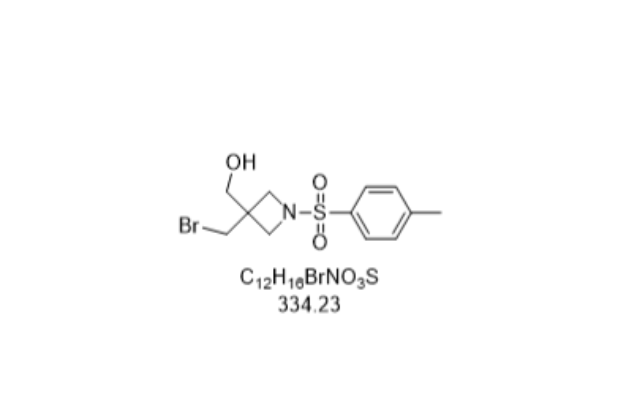
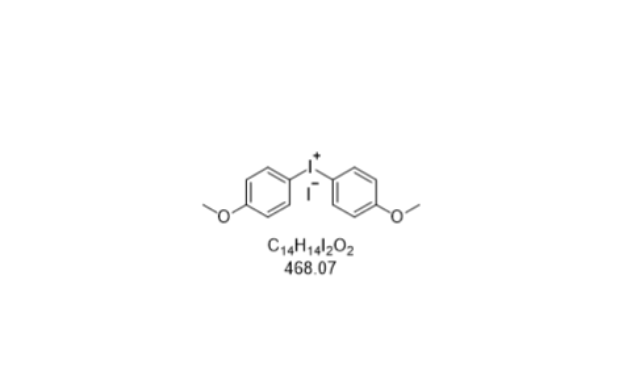
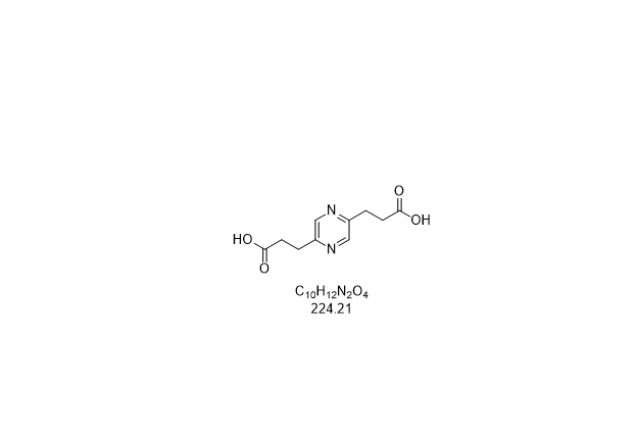
- 石家庄4-溴-2-甲基茚 03-04
- 2-碘-5-溴嘧啶厂家供货 03-04
- N-(2-(二乙基氨基)乙基)-2 03-04
- 常州磺酰二咪唑 03-04
- (4-溴苯基)乙胺现价 03-04
- 长沙4-苯基-2-甲基茚 03-04
- 郑州紫杉醇侧链中间体(3R 03-03
- 福州对溴苯腈 03-03
- 无锡4-对叔丁基苯基-2-甲基茚 03-03
- 太原2-溴-1 03-03